|
home | purchase | services | contact | about |

|

|

|

|

|

|

|

|

|
|
Applications and Protocols |  |
IntroductionThe Applications and Protocols section contains an overview of various antibody and nucleic acid assays and reaction types that may be performed using the Parallume technology platform. Proteins and antibodies may be bound to the Parallume encoded beads covalently or electrostatically using carboxylated or amino-derivitized beads, respectively. Likewise, nucleic acids may be electrostatically bound or amino-derivitized oligonucleotides used for covalent attached to the beads. The Applications section below discusses general assay types for which the Parallume beads may be used. The Protocols section covers the materials and methods used for attaching DNA or antibodies to the beads and instructions for performing various common assays. Information on operating the MARS instrument, and the features of the MARSoft acquisition and analysis software, may be found in the MARS User Manual. A. ApplicationsThe Applications below are examples of the type of multiplexed reactions and assays that may be performed with the Parallume encoded beads. Please see the Protocols section below for details concerning the materials and methods. The sampling of antibody and DNA assays shown here provides a view of the type, number and variety of what reaction types and assays may be amenable to multiplexing with the Parallume encoding technology. Please Contact us to ask any questions you might have about experimental multiplex design or any other requirements. A.1 Antibody and Protein AssaysThere are a large number of protein and antibody assay types which may be performed using the Parallume beads some of which are illustrated in Fig. 1. Typically, a capture antibody (Ab1 in Fig. 1A) is bound to the bead either covalently (using the amino groups on Ab1 and a carboxylated bead surface) or electrostatically (using amino-derivitized beads). The agarose beads are completely stable toward the EDC/NHS method for coupling amines to carboxylic acids.  Fig. 1 Any antigen-antibody or protein-protein interaction maybe studied in a multiplexed fashion using Parallume encoded beads. The antibodies or proteins may be attached to the beads via electrostatic or covalent bonding. After the multicomponent antigen target mixture is selected, a different primary capture antibody against each antigen is attached to its own unique Parallume encoded bead set (A), all bead sets pooled together and treated with the target mixture (B). To detect the captured antigen a dye-labeled detection antibody is bound either (a) directly to the antigen (C) or (b) to an antibody which is bound to the antigen (D). The beads are first exposed to the target antigen which bonds to the bead-bound capture antibody (Fig. 1B). The amount of captured antigen is determined using a reaction that directly or indirectly stains the beads to indicate antigen level. The antigen may be visualized by either (a) treatment with a labeled secondary antibody (Ab2 Fig. 1C) or treatment with a secondary antibody followed by treatment with a third staining antibody as shown as Ab3 in Fig. 1D. A.2 Nucleic Acid AssaysWhile a very large number of nucleic acid assays may be performed on encoded Parallume beads, there are several important and unique assays which derive from the exceptional thermal stability of the agarose Parallume beads. The thermal stability allows the beads to be present during thermal cycling under PCR conditions or to be autoclaved. The numerous nucleic acid assays which may be multiplexed include, but are not limited to, hybridization, clonal on-bead amplification, ligation, primer extension, common primer mediated amplifications, detection of unlabeled target DNA sequences, capture of transcription factors, capture of RNA sequences and many other assays. It is important to note that diffusion rates of nucleic acids and other reagents in and out of the highly porous Parallume agarose beads allows the probe-target binding reaction to reach steady state much faster than the corresponding assays on planar substrates. For example, hybridization reactions take place in minutes rather than hours as shown in Fig. 2.  Fig. 2 When a 1µM target solution of a Cy-dyed 26 nt target is stirred with its complementary capture probe bound to Parallume beads the hybridization reaction reached steady state in less than 2 minutes (top), while a 1nM 100nt target requires ~20 minutes (bottom). A.2.1 Multiplex Hybridization AssaysMultiplex hybridization is one of the most common methods used to distinguish and quantitate PCR products, or other mixed nucleic acid samples, in multiplexed assays. In the case of the Parallume assays, the desired target is captured onto its specific probe which resides on a bead with a unique Parallume optical code. To determine the amount of all targets present in the multicomponent mixture to be analyzed, the beads containing appropriate capture probe are simply pooled with the target mixture, incubated, washed and imaged to provide target levels. Pooled targets may be detected by multiplex hybridization in several different ways. If the target sequence can be directly dye-labeled, then the labeled target may be measured directly as schematically illustrated in Fig. 3. 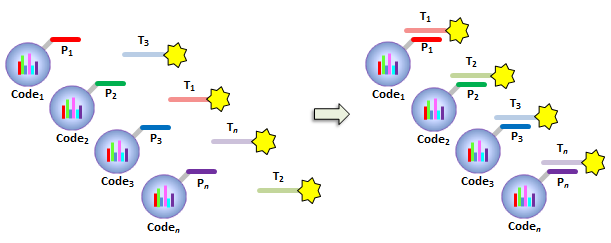 Fig. 3 - PCR products or any mixture of target nucleic acids (T) may be detected by multiplex hybridization assays by placing each capture probe (P) onto a bead with a unique Parallume optical code and combining the pooled beads with the multicomponent target mixture. In this example, the target sequence has been dye-labeled but methods to detect unlabeled sequences are shown below in Fig. 4. The target fluorescent signal level on a given bead code is related to the target concentration in the analyzed sample. If it is not possible to directly label the target, then it is possible to detect an unlabeled target by either of the methods shown in Fig. 4. In one method (Fig. 4A), a third reporter sequence, which is complementary to the target and conterminous to the probe, is used to determine the reporter amount bound to the probe. The reporter may be dye-labeled itself (Fig. 4A) or possess a biotin for SAPE staining. In another method (Fig. 4B), the familiar Stem-Loop structure provides both the capture probe and reporter functionality in a single sequence and eliminates the need to wash the beads free of any unbound reporter. Unlike polystyrene beads, the Parallume beads are perfectly stable toward denaturing double strand DNA at 98°C or hybridizing at any temperature. 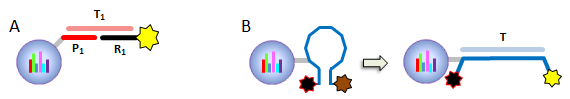 Fig. 4 - If a dye-labeled target is not available, it is possible to detect and quantify unlabeled target DNA using either (A) a tripartite Probe-Target-Reporter assemblage where the reporter is complementary to the target and conterminous to the probe or (B) the famous and familiar Stem-Loop structure. The Stem-Loop structure obviates the need for washing the beads. An example of a 20-plex hybridization is shown in Fig. 5. 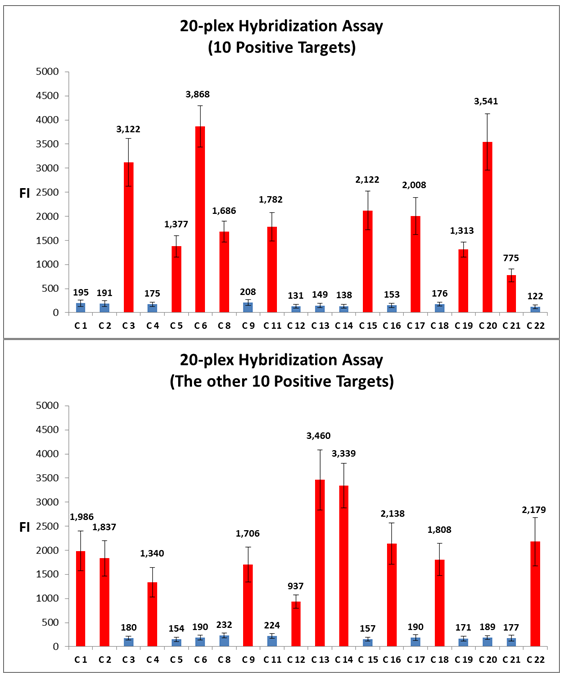 Fig. 5 - An example of a 20-plex hybridization. There are 20 pooled Parallume bead codes (C) each with a capture probe against one of 20 target sequences. When the 20 bead codes/probes are mixed with 10 of the 20 targets, 10 of twenty beads show a positive signal (top). In a separate experiment, the same 20 codes/probes are treated with the remaining 10 targets and the remaining 10 of twenty beads show a positive signal (bottom). Note low and uniform background for non-homologous sequences. A.2.2 PCR and Other Thermal Amplification AssaysWhile any type of assay may be performed on the Parallume beads, the ability to use the thermally stable cross-linked agarose beads for PCR and related thermal applications is of particular importance. The Parallume beads may be present throughout the thermal cycling and act either passively, by detecting PCR products via multiplex hybridization detection, or participate directly in the amplification by primer extension on the bead surface. In either case, unlike polystyrene beads, the thermally stable beads eliminate the need to open the vessel for detection after thermal amplification. A.2.2.1 Primer ExtensionAs schematically illustrated in Fig. 6, it is possible to extend bead-bound primers on the surface of Parallume encoded beads. We show below that different primers on different beads all extend at approximately the same rate and to the same extent. 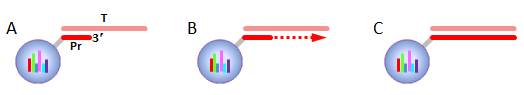 Fig. 6 - Bead-bound primers (Pr) may be easily extended using standard methods. The bead-bound primer (A) anneals to the target (T), undergoes primer extension (B) to give the extended double strand product (C). A.2.2.2 Bridge PCR (BPCR) and Clonal AmplificationWhen the two primers required to amplify a target are bound to the surface of the same bead and target is added, the two primers are extended in the fashion illustrated schematically in Fig. 7. The ability to amplify one particular sequence on one bead (Fig. 7) allows this type of dual primer Parallume bead to be used in a manner similar to the clonal amplification within water droplets within water in oil emulsions. 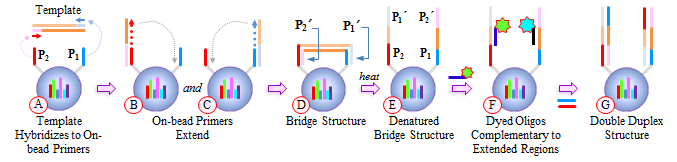 Fig. 7 - An example of BPCR. When two bead-bound primers (A) complementary to the target extend (B and C) it possible to form a bridge structure as shown in (D and E). The data shown in Fig. 8 demonstrates that during multiplex BPCR that all primers on all beads extend at the same rate and to the same extent. An example of a 10-plex BPCR on Parallume beads is shown in Fig. 8 where it can be seen that all ten primer pairs extended at the same rate and to approximately the same extent. These results again demonstrate the complete stability of the Parallume beads toward thermal cycling under PCR conditions. 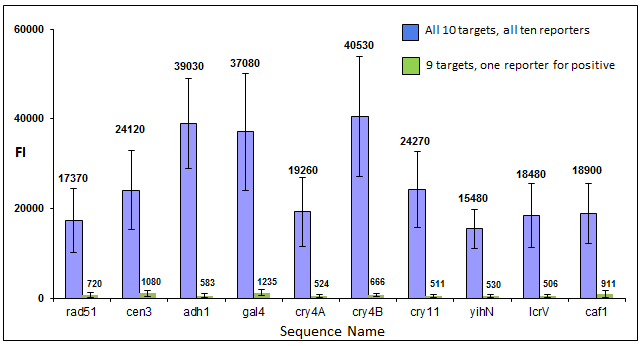 Fig. 8 - Using the Probe-Target-Reporter (PTR) assemblage (Fig. 4A), ten Parallume encoded beads sets each containing primer pairs for 10 target sequences are thermally cycled under PCR conditions and stained with 10 specific reporters which are complementary to sequences generated by extension only (blue bars) as schematically illustrated in Fig. 7F. For the negative control reactions, 10 separate 10-plex reactions were preformed and each in each 10-plex BPCR one of the 10 targets was omitted but the stain for that extended region was present. These results show that each isolated primer pair generated BPCR product at a constant rate, and to largely the same extent, on all beads. This on-bead BPCR technique may be used as one component in a scheme that amplifies, without any bias, arbitrarily large number of target sequences from a sample as shown below in Fig. 9. A.2.2.3 Common Primer Mediated Amplification SchemesThere are a variety of protocols whereby a large number of different targets may be amplified in solution by PCR but the methods which amplify different sequences which all share a common region can sometimes amplify all PCR products with bias, i.e. no one PCR product is amplified more efficiently than another. This is a result of the fact that there is only a single primer pair in solution. 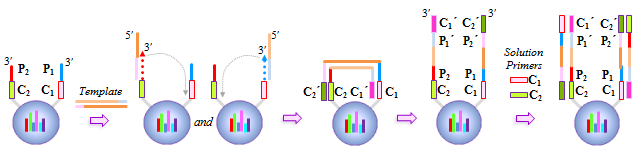 Fig. 9 - When the multiplex BPCR amplification shown in Figs. 7 and 8 is performed with the addition of two commons regions (C1 and C2) on every unique primer pair (P1 and P2) on every bead, and two solution phase primers with sequences C1 and C2 are added, a “double duplex” structure is generated (far right). Denaturation of the double duplex provides input for a solution phase mediated amplification as depicted in Fig. 10. When the BPCR amplification is carried out as depicted in Fig. 7, but with shared common sequences among all beads on each of the two unique primer sequences flanking the target sequence to be amplified (Fig. 9), every extended BPCR product on every bead now contains the complements to the two common regions at their 3´ end (Fig. 9). As schematically illustrated in Fig. 9, a double duplex structure is generated in the presence of the two primers C1 and C2 which are the same common sequences which were originally conterminous to the bead-bound PCR primers. When denaturation of the double duplex structure occurs in the presence of soluble C1 and C2 solution phase PCR ensues (Fig. 9). Since all bead-bound amplicons on all beads have the same common priming regions, the single primer pair in solution should amplify all the different sequences at the same rate without PCR bias. 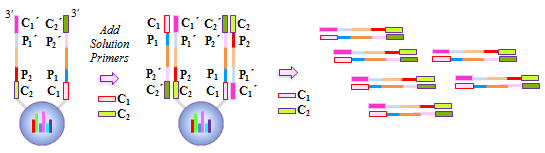 Fig. 10 - Denaturation of the double duplex (center) provides selective input for the common primer mediated solution phase amplification. Once formed on the double duplex, the sequence are amplified along with all other amplicons containing the same common region without any PCR amplification bias as there is only one primer pair in solution. A.3 Small Molecule and Other Affinity ReactionsSince (a) the pores in the Parallume encoded agarose beads are large enough to accommodate proteins such as IgG and streptavidinphycoerythrin, (b) small molecules can diffuse very rapidly within the pores, (c) there a variety of different surface linker chemistries available and (d) the agarose host material is stable toward heat and many common reagents, the Parallume beads may serve as ideal screening platform for small molecules or biological materials. For example as depicted in Fig. 11, a binding reaction between two components may be studied by attaching one of the species to the Parallume encoded bead and its binding partner in solution is dye labeled. The dye signal on any given Parallume code is indicative of the amount of dye labeled target bound to that capture probe.  Fig. 11 - In order to use the Parallume encoded beads for screening applications, the probe or the target may be attached to the bead and its solution phase counterpart dye labeled. A.4 Multiplex Cell and Tissue StainingWe have developed a process whereby Parallume nanoparticles can be rendered highly dispersible and with surface carboxylate linker groups suitable for attachment of proteins, antibodies, antigens or DNA. Please contact Parallume to discuss the type of nanoparticles that you require for your experiments. B. ProtocolsB.1 General Bead Handling ProceduresThese procedures are suitable for either (a) covalently attaching a capture antibody to the beads or (b) covalently attaching a recombinant protein to the beads using the well-known EDC/NHS protocol. Smaller peptides often bind spontaneously to the beads without chemical crosslinking but larger proteins and all antibodies require the crosslinking procedure given here to covalently link them to the beads. 1) The virgin Parallume beads, which have never been treated with any proteins, surfactants, detergents, cell extracts or nucleic acids, i.e., as received from Parallel, are reactive and may adhere to the walls of the pipet tip, reaction tube or clump together. If the underivitized beads cling to the pipet tip when using your buffer, the use of commercially available “low adhesion” pipet tips is recommended. If the beads clump or adhere to the tube during protein conjugation or coating, just continue with the protocol. Once the buffer contains surfactant, proteins, antibodies, etc., or the PBST used here, in subsequent steps of the assay, the beads will no longer adhere to surfaces and should become free floating once again. 2) Stirring means agitation on an orbital plate shaker (suggested: 5 mm orbit). The speed of shaking required to best suspend the beads depends on the size and shape of the tube and the amount of beads used. For 0.1–50 μL of beads in 10-1000 μL of buffer, a 1.5 mL round bottom centrifuge tube (Eppendorf) is shaken at 800 RPM, a 1.5 μL conical-bottom tube at 1200 RPM and a 200μL PCR tube at >1700 RPM. In general, the beads are easiest to stir in the round bottom tubes. Bead volumes of several μL or larger may be conveniently handled in a 1.5 mL Pall Nanosep with a 500 μL 0.45μm filter insert. Visually inspect the beads when on the plate shaker to make sure the liquid vortex rises up out of the bottom of tube and coats the size of the tube - but not so fast that liquid adheres uniformly to the entire tube wall from centrifugal force. Rotation at too fast or too slow a speed results in less efficient mixing and potentially compromised detection limits. If the beads are not mixed as vigorously as required, the kinetic diffusion of material in and out of the porous beads, as well as the detection limits, could suffer. 3) To obtain more uniformity among beads during a reaction, do not add a concentrated solution of a reagent to the beads without agitation, but rather immediately aspirate the solution in and out of the pipet tip as rapidly as possible in order to expose all beads as quickly as possible to the same concentration of reagent at the same time. 4) Unless otherwise specified, washing means stirring for 5 minutes on the plate shaker 5) For isolation, the beads may be quickly centrifuged on a small centrifuge, pulled down with a magnet if magnetic beads are used or just allowed to settle by gravity. 6) Settled beads refer to beads that have been allowed to settle for at least 10 minutes or centrifuged. One μL of settled beads is about 1 mg of beads and there are approximately 1500-3000 beads/μL. In general, ten or more beads are needed per assay depending on the particular assay. 7) To measure a small volume of beads, pipet a volume of beads into the appropriate dilution volume, quickly aspirate the beads in and out of the pipet tip to completely and uniformly suspend them then transfer desired volume of suspended (diluted) beads. Accurate aliquotting requires a uniform and homogeneous bead suspension. 8) Store bare, unused Parallume beads at room temperature or 4 °C in 0.03% NaN3. Beads are always supplied in 0.03% NaN3 unless otherwise specified. B.2 Protocol for Conjugating Antibodies or Recombinant Proteins to Encoded Agarose Beads
B.3 Protocol for Blocking Antigen or Antibody-derivitized BeadsWhen using an anti-species reporter stain, it is best to use serum from that species for blocking if possible. For example, if the detection antibody is a monoclonal mouse antibody, block with normal rabbit serum if the anti-species antibody reporter stain is rabbit anti-mouse. Use 1-5 % vol:vol normal serum:PBST for blocking.
B.4 Protocol for Pre-clearing Human Serum for Immunoassays on Agarose BeadsIf human serum is part of the assay, there appears to be a human antibody against agarose present in all human serum that must be removed from the serum before any assay is performed.
B.5 Protocol for ELISA or Antibody CaptureDepending on whether an antigen (antigen down assay) or antibody (ELISA) was coupled to the beads as the capture probe, the following protocol is used to capture and stain the target antibody or antigen, respectively.
Target Staining If the target is not labeled, then the bead-bound target or detection antibody must be stained with a reporter antibody.B.6 Protocol for Coupling Amine-Modified Oligonucleotides to Carboxylated Agarose Beads
(1) It is recommended that the amino-oligonucleotide contain a linker group of six (-CH22O-) PEG linker groups, i.e., (-CH2CH2O-)6, between the amino functionality and the oligo itself. A hexamethylene (-CH2-)6 linker may be used but is shorter and hydrophobic. (2)There are ~1-2 x 103 beads/mg and ~10 or more beads are required for each sample analysis.B.7 Protocol for Hybridization to Bead-bound Oligonucleotide Capture Probes
Pre-Hybridization Blocking Procedure
|
Copyright © 2000-2015 Parallel Synthesis Technologies, Inc. |